
22q11.2缺失综合征(其也由其他几个名称,下面列出已知的)是由一种病症的缺失一小段22号染色体。缺失发生在染色体中部附近的q11.2位置。
22q11.2缺失综合征有许多可能的症状和体征,几乎可以影响身体的任何部位。这种综合征的特征差异很大,甚至在同一家庭的受影响成员中也是如此。常见的症状和体征包括心脏异常,常常从出生,在口腔顶部的开口(目前裂腭),和独特的面部特征。患有22q11.2缺失综合征的人经常会出现由免疫系统问题引起的反复感染,并且一些患有自身免疫性疾病,例如类风湿性关节炎和格雷夫斯病,其中免疫系统攻击身体自身的组织和器官。受影响的个体也可能有呼吸问题,肾脏异常,血液中钙含量低(可导致癫痫发作),血小板减少(血小板减少),严重的喂养困难,胃肠道问题和听力丧失。骨骼差异是可能的,包括轻度身材矮小,脊柱骨骼异常较少。
许多患有22q11.2缺失综合征的儿童发育迟缓,包括生长迟缓和言语发育以及学习障碍。在以后的生活中,他们患精神分裂症,抑郁症,焦虑症和双相情感障碍等精神疾病的风险增加。此外,受影响的儿童比没有22q11.2缺失综合征的儿童更有可能患有注意力缺陷多动障碍(ADHD)和影响交流和社交互动的自闭症谱系障碍等发育状况。
由于22q11.2缺失综合征的体征和症状是如此多样,因此不同的特征分组一度被描述为单独的条件。医生将这些病症命名为DiGeorge综合征,velocardiofacial综合征(也称为Shprintzen综合征)和conotruncal异常面部综合症。此外,一些22q11.2缺失的儿童被诊断为常染色体显性形式的Opitz G / BBB综合征和Cayler贲门综合征。一旦确定了这些疾病的遗传基础,医生就确定它们都是单一综合征的一部分,并伴有许多可能的体征和症状。为避免混淆,这种情况通常称为22q11.2缺失综合征,这是一种基于其潜在遗传原因的描述。
22q11.2缺失综合征发病率
22q11.2缺失综合征影响估计每4,000人中就有 1人。然而,这种情况实际上可能比这个估计更常见,因为医生和研究人员怀疑它由于其可变特征而被诊断不足。患有轻度体征和症状的人可能无法确定病情,或者可能将其误认为具有重叠特征的其他疾病。
22q11.2缺失综合征发病原因
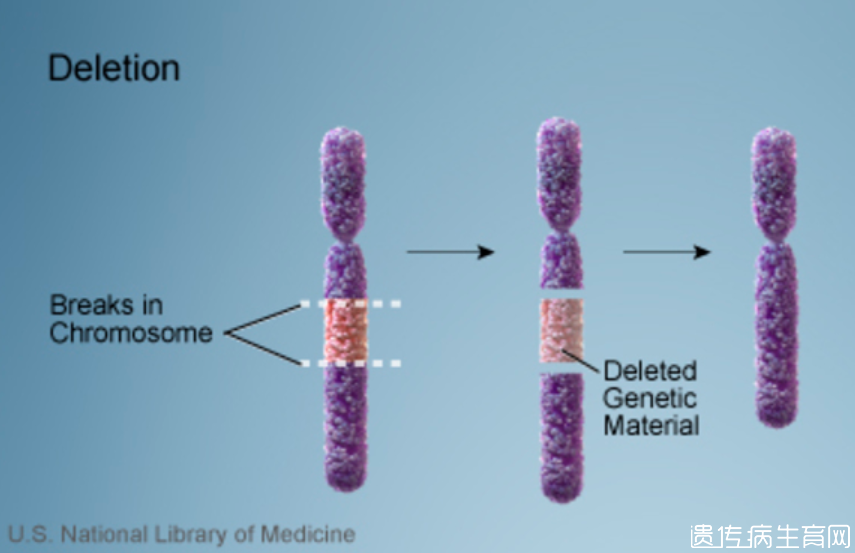
大多数患有22q11.2缺失综合征的人缺少一个序列大约300万个DNA构建块(碱基对)在每个细胞中的22号染色体上。该区域含有30至40个基因,其中许多基因尚未得到很好的表征。一小部分受影响的个体在同一地区的缺失较短。这种情况被描述为一种连续的基因缺失综合症,因为它是由于许多基因缺失而导致的。
研究人员正在努力确定导致22q11.2缺失综合征特征的所有基因。他们已经确定,在一个特定基因的损失22号染色体,TBX1,可能是负责许多综合征的特征性体征(如心脏缺陷,裂腭,独特的面部特征,听力损失和低钙水平)。一些研究表明,删除该基因也可能导致行为问题。在22号染色体的同一区域中另一个基因COMT的丢失也可能有助于解释行为问题和精神疾病的风险增加。缺失区域中额外基因的丢失可能有助于22q11.2缺失综合征的不同特征。
22q11.2缺失综合征发病率
继承22q11.2缺失综合征被认为是常染色体显性因为一个缺失在每个细胞中的22号染色体的一个拷贝中足以引起该病症。然而,大多数22q11.2缺失综合征病例并未遗传。缺失最常发生在生殖细胞(卵子或精子)形成期间或胎儿早期发育期间的随机事件中。受影响的人通常没有家庭疾病的病史,尽管他们可以将病情传给孩子。在大约10%的病例中,患有此病症的人继承了删除在染色体22从父。在继承的案件中,其他家庭成员也可能受到影响。

